

La Sindrome di Rett è una rara malattia del neurosviluppo causata da una mutazione de novo a carico del gene MeCP2. La sindrome inizia a manifestarsi dal sesto mese di età ed è caratterizzata da una regressione delle capacità precedentemente acquisite (ad esempio a livello del linguaggio). I sintomi comprendono:
- disprassia o atassia (incapacità o alterazione nello svolgimento dei movimenti)
- perdita della capacità di comunicazione
- irritabilità
- stereotipie (la più nota è l’hand-washing, uno sfregamento continuo simile al lavaggio delle mani)
- crisi epilettiche
- scoliosi
- disturbi respiratori (ad esempio apnee ed iperventilazione)
- battito cardiaco irregolare
- costipazione
Questi sintomi possono insorgere in momenti differenti e tendono a peggiorare nelle fasi più tardive della sindrome (intorno ai 20 anni).
Basi genetiche della sindrome:
Le femmine di mammifero possiedono in ciascuna cellula due copie del cromosoma X, una di origine materna, l’altra di origine paterna. Pertanto in ogni cellula uno dei due cromosomi X viene silenziato, evitando una sovraespressione dei prodotti delle due X. La disattivazione è un fenomeno casuale, quindi può colpire ciascun cromosoma X con uguale probabilità. Questo fenomeno biologico prende il nome di mosaicismo, poiché le femmine appaiono come un mosaico tra i caratteri X-linked (portati sul cromosoma X) di derivazione materna e paterna.
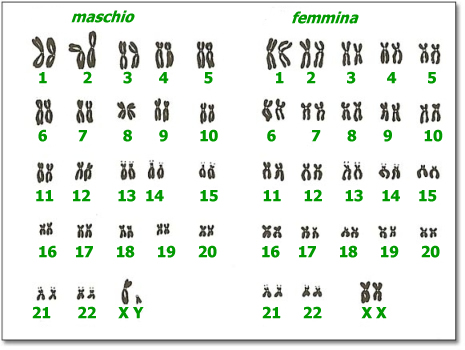
Il gene MeCP2 è presente a livello del cromosoma X (la Sindrome di Rett è X-linked), pertanto le bambine colpite dalla sindrome mostrano un fenotipo differente, in base al numero di cellule in cui è espressa la X con MeCP2 mutato, rispetto alle cellule in cui è espressa la X sana. Ne deriva che la gravità della sintomatologia, i tempi di insorgenza e lo stesso spettro di sintomi manifestato differisce da bambina a bambina. Per quanto riguarda invece i bambini maschi, questi hanno un solo cromosoma X, perciò quando la mutazione è presente la sindrome si manifesta precocemente e con una sintomatologia severa (molti bambini muoiono prima della nascita o entro i primi mesi di vita). Per questo motivo, quando si parla di Sindrome di Rett, i pazienti sono principalmente bambine e ragazze.
Anche il tipo di mutazione a carico di MeCP2 determina una diversa gravità dei sintomi ed influenza l’età di inizio della regressione.
Aspetti neurobiologici:
La mutazione a carico del gene MeCP2 comporta alterazioni microscopiche nel Sistema Nervoso. Infatti, a livello dell’arborizzazione dendritica è possibile notare come gli individui affetti dalla Sindrome di Rett presentino un significativo impoverimento delle ramificazioni e dunque una perdita di sinapsi (se non sai cos’è una sinapsi, clicca qui).
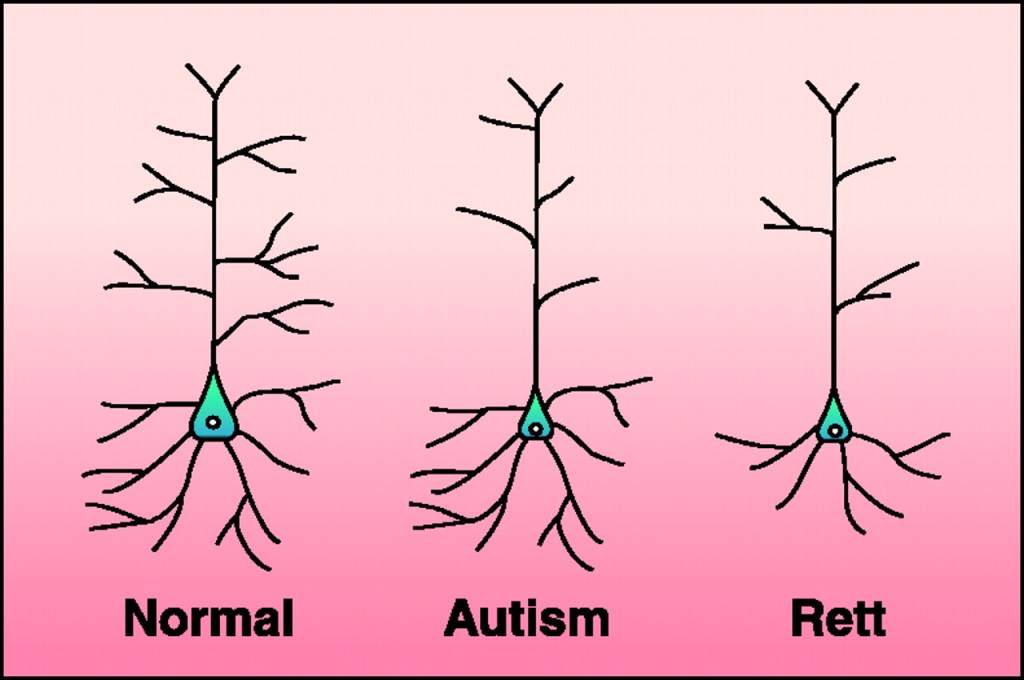
Questa perdita di spine dendritiche riflette la regressione tipica delle capacità precedentemente apprese ed è dovuta ad una ridotta produzione di BDNF (Brain Derived Neurotrophic Factor), una proteina coinvolta nel neurosviluppo, nei processi di neurogenesi, di mantenimento dell’efficacia sinaptica e di neuroplasticità (se vuoi approfondire l’argomento, clicca qui).
Ipotesi di trattamento:
Anche se attualmente non esiste una cura per la Sindrome di Rett, negli anni sono state avviate numerose ricerche sul modello murino per individuare possibili opzioni di trattamento. Molti studi si sono concentrati sulla somministrazione di specifici fattori molecolari (come il BDNF), mentre altri hanno valutato le risposte a protocolli di arricchimento ambientale (clicca qui per approfondire). In particolare, è stato visto come la permanenza in ambiente arricchito già dai primi giorni di vita possa migliorare e in alcuni casi persino recuperare i deficit cognitivi e motori nel topo (Lonetti et al., 2010; Kondo et al., 2016). Questi risultati erano anche associati ad un aumento dei livelli di BDNF, raggiungendo i livelli degli animali sani.
Sulla base di queste evidenze, Down e collaboratori hanno esaminato le risposte ad un protocollo di ambiente arricchito in un gruppo di bambine affette da Sindrome di Rett.
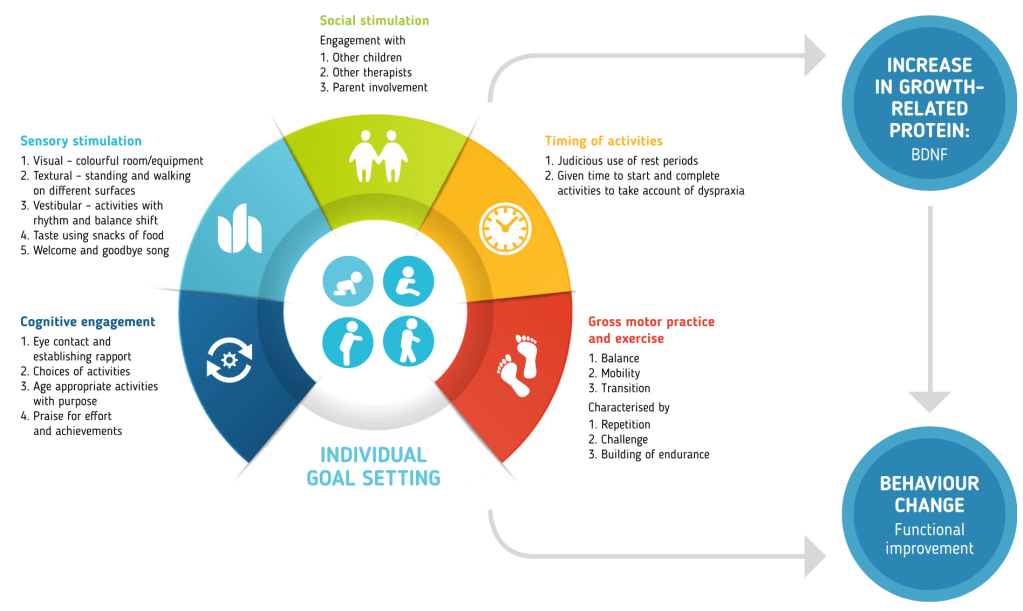
Le bambine coinvolte in questo studio avevano un’età media di 3 anni e possedevano tra di loro diverse mutazioni a carico del gene MeCP2, pertanto non condividevano i medesimi livelli di deficit cognitivi e motori. L’intervento prevedeva l’esposizione ad un ambiente ricco di stimoli sensoriali e motori, in cui alle bambine venivano proposte principalmente attività di equilibrio e deambulazione, basate sull’apprendimento motorio. Ognuna di queste attività era svolta assieme almeno ad un terapista ed era integrata con esperienze visive (colori, giocattoli), uditive (canto), gustative (piccoli snack), vestibolari (spostamento dell’equilibrio) o tattili (differenti superfici su cui camminare o gattonare). Il protocollo prevedeva lo svolgimento di queste attività per 2 o 3 ore, 6 mattine a settimana, per un periodo di 6 mesi, durante il quale sono stati valutati eventuali differenze a livello delle capacità motorie e della quantità di BDNF.
L’apprendimento motorio e l’esercizio, integrati con esperienze sociali e somatosensoriali, hanno avuto effetti positivi sulle funzioni motorie delle bambine, in particolare per coloro che avevano una forma clinicamente più lieve della sindrome. Anche i livelli di BDNF hanno subito un incremento, rispetto ai valori iniziali, in accordo con studi precedenti che mostravano come nella popolazione generale l’esercizio aerobico fosse correlato ad un aumento del BDNF (Huang et al., 2014). Problematiche inerenti alla salute generale non hanno mostrato risentire delle attività svolte, indicando che è possibile proporre dei protocolli di questo tipo per la Sindrome di Rett, in condizioni di sicurezza e senza compromettere lo stato di salute dei pazienti.
Questo studio quindi suggerisce che l’ambiente arricchito abbia benefici per i bambini affetti da questa sindrome e che interventi comportamentali di questo tipo costituiscano un importante complemento evidence-based nella gestione della malattia. Ulteriori studi dovranno essere svolti per valutare se protocolli protratti per oltre 6 mesi possano portare un contributo aggiuntivo, specialmente alle bambine che sviluppano una forma più grave della sindrome.
Potrebbe anche essere utile considerare delle strategie che comprendano interventi di arricchimento ambientale, affiancati alle terapie farmacologiche.
Bibliografia:
- Downs J., Rodger J., Li C., Tan X., Hu N., Wong K., De Klerk N., Leonard H. (2018) Environmental enrichment intervention for Rett syndrome: an individually randomised stepped wedge trial. Orphanet Journal of Rare Diseases (2018) 13:3
- Huang T., Larsen K. T., Ried-Larsen M., Moller N.C., Anderson L.B. (2014) The effects of physical activity and exercise on brain-derived neurotrophic factor in healthy humans: A review. Scandinavian Journal of Medicine and Science in Sports 24:1–10
- Kondo M. A., Gray L. J., Pelka G. J., Leang S., Christodoulou J., Tam P. P. L., Hannan A. J. (2016) Affective Dysfunction in a Mouse Model of Rett Syndrome: Therapeutic Effects of Environmental Stimulation and Physical Activity. Developmental Neurobiology 76(2):209-24
- Lonetti G., Angelucci A., Morando L., Boggio E. M., Giustetto M., Pizzorusso T. (2010) Early Environmental Enrichment Moderates the Behavioral and Synaptic Phenotype of MeCP2 Null Mice. Biological Psychiatry 67(2):657–65
- Zoghbi H. Y. (2003) Postnatal Neurodevelopmental Disorders: Meeting at the Synapse? Scienze 302(5646):826-30
Grazie per aver letto questo articolo!
Nota bene: la fotografia non è stata scattata durante lo studio citato, né vuole esserne rappresentativa.

1 pensiero su “Effetti dell’ambiente arricchito nella Sindrome di Rett”